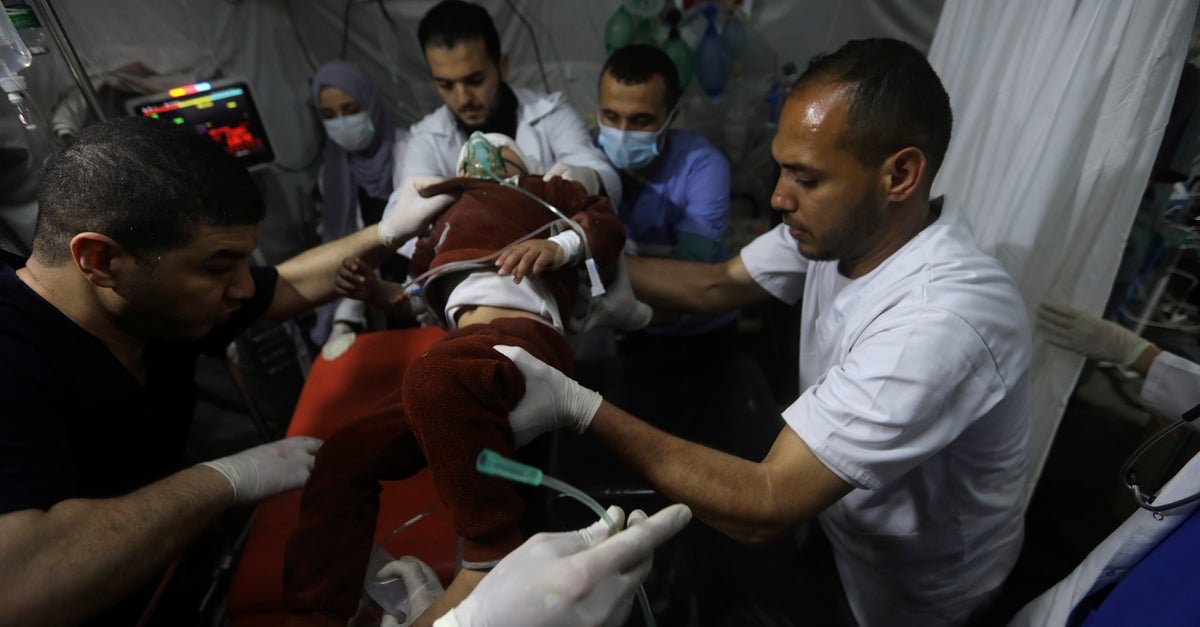

Dans une petite étude, le modulateur allostérique de la pyruvate kinase (PK) mitapivat (Pyrukynd) a été efficace dans le traitement de l’anémie hémolytique chez les patients atteints d’α-thalassémie et de β-thalassémie qui ne dépendent pas des transfusions.
Selon Kevin HM Kuo, MD, de l’Université de Toronto, 16 des 20 patients (80 %) ont présenté une réponse à l’hémoglobine – définie comme une augmentation de 1,0 g/dL ou plus de la concentration d’hémoglobine par rapport au départ – lorsqu’ils ont été traités avec le mitapivat. en Ontario, et ses collègues.
L’étude « établit une preuve de concept selon laquelle l’activation de la PK par l’agent oral mitapivat améliore l’anémie chez les patients atteints de [non-transfusion-dependent] α-thalassémie et β-thalassémie », ont écrit Kuo et ses collègues dans Le Lancetajoutant que les résultats d’efficacité et d’innocuité “fournissent une justification pour la poursuite des recherches sur le mitapivat pour le traitement de l’α-thalassémie et de la β-thalassémie”.
Au total, 15 des patients avaient une β-thalassémie et cinq une α-thalassémie. Tous les cinq de ces derniers avaient une réponse à l’hémoglobine, ainsi que 11 des 15 (73%) avec la β-thalassémie.
Dix-neuf (95 %) patients ont terminé la période de traitement principale au cours de laquelle ils ont reçu une dose initiale de 50 mg de mitapivat par voie orale deux fois par jour pendant les 6 premières semaines de la période d’étude principale de 24 semaines. Si le médicament était bien toléré, la dose était augmentée à 100 mg deux fois par jour.
Un patient qui a eu une β-thalassémie a arrêté après la semaine 4 en raison d’un événement indésirable lié au traitement (TEAE) sans rapport avec le mitapivat. Tous les patients, à l’exception de celui qui a arrêté prématurément, ont reçu une dose accrue de mitapivat à la semaine 6.
Sur les 16 patients avec une réponse primaire d’hémoglobine, 13 ont eu une réponse en recevant 50 mg deux fois par jour au cours des 6 premières semaines, et trois ont eu une réponse en recevant 100 mg deux fois par jour au cours des 6 semaines suivantes. De plus, une réponse retardée de l’hémoglobine (après la semaine 12 et après l’augmentation de la dose à 100 mg deux fois par jour) a été observée chez deux patients, tous deux atteints de β-thalassémie.
Dans un commentaire accompagnant l’étude, Antonis Kattamis, MD, de l’École de médecine de l’Université nationale et kapodistrienne d’Athènes, a écrit que même si l’étude était “limitée à la fois par le nombre de patients participants et la durée de l’observation”, les “résultats sont indéniablement positif.”
Il a soulevé plusieurs questions, dont celle d’une dose optimale, qui, selon lui, nécessite une évaluation plus approfondie.
Les différences de variation moyenne de la concentration d’hémoglobine par rapport au départ après l’augmentation de la dose à la semaine 6 suggèrent “qu’il n’y a pas de corrélation dose-efficacité linéaire”, a écrit Kattamis.
“Bien que le profil de sécurité semble similaire entre les doses de 50 mg et 100 mg, des doses accrues pourraient ne pas être nécessaires pour la réponse et pourraient affecter l’observance à long terme”, a-t-il poursuivi. “De plus, si le médicament est approuvé, le traitement à une dose accrue sera associé à des coûts accrus, qui pourraient être difficiles à couvrir par différents systèmes de santé, en particulier dans les pays à faibles ressources, où résident de nombreux patients atteints de thalassémie.”
Kuo et ses co-auteurs ont noté que le profil d’innocuité du mitapivat était similaire à celui observé dans les études précédentes. Un total de 17 patients ont eu un TEAE au cours de la période principale, dont 13 ont été considérés comme liés au traitement.
Les EIAT les plus fréquemment signalés (survenant chez ≥ 10 % des patients) étaient l’insomnie initiale, les étourdissements et les céphalées, avec des durées médianes de 34 jours pour l’insomnie initiale, de 2 jours pour les étourdissements et de 14,5 jours pour les céphalées. Des EIAT de grade 3 ou pire ont été signalés chez cinq patients et comprenaient une insomnie initiale, une arthralgie, une insuffisance rénale, une anémie et des vertiges positionnels. L’insomnie initiale était le seul EIAT de grade 3 ou pire considéré comme lié au traitement.
Les chercheurs ont noté que la thalassémie non dépendante des transfusions revêt une importance mondiale croissante en raison de la migration d’individus provenant de régions à forte incidence de la maladie, comme la Méditerranée et l’Asie du Sud-Est, vers des régions où l’incidence est généralement faible, comme comme les États-Unis et le Royaume-Uni En outre, selon les auteurs, la thalassémie non dépendante des transfusions est l’une des hémoglobinopathies les plus fréquemment identifiées dans certains programmes de dépistage des nouveau-nés.
Bien que des transfusions sanguines régulières ne soient pas nécessaires pour la survie de ces patients, ils “ont toujours un fardeau important de comorbidités et de complications liées à la maladie et une qualité de vie réduite”, ont déclaré les enquêteurs.
L’étude de phase II en ouvert a été menée sur quatre sites cliniques universitaires en Amérique du Nord et au Royaume-Uni entre décembre 2018 et février 2020. Les patients étaient éligibles s’ils étaient âgés de 18 ans ou plus, avaient une thalassémie non dépendante des transfusions (y compris la β-thalassémie avec ou sans mutations du gène α-globine, hémoglobine E β-thalassémie ou α-thalassémie), et une concentration d’hémoglobine de base de 10,0 g/dL ou moins.
L’âge médian des patients au départ était de 44 ans et la majorité (75 %) étaient des femmes. Tous les patients atteints d’α-thalassémie étaient de race asiatique et un tiers des patients atteints de β-thalassémie ont été identifiés comme asiatiques.
Les limites de l’étude, selon les chercheurs, comprenaient la conception ouverte à un seul bras sans groupe témoin et le petit nombre de participants. De plus, les chercheurs n’ont pas effectué de tests d’hypothèse sur les changements dans les marqueurs de l’hémolyse et de l’érythropoïétine, et l’augmentation séquentielle de la dose a empêché des inférences définitives concernant les effets dose-dépendants.
-
Mike Bassett est un rédacteur qui se concentre sur l’oncologie et l’hématologie. Il est basé dans le Massachusetts.
Divulgations
L’étude a été financée par Agios Pharmaceuticals.
Kuo a signalé des relations financières avec Agios, Alexion, Apellis, Bluebird Bio, Celgene, Forma, Pfizer, Novartis et Bioverativ/Sanofi/Sangamo ; les co-auteurs ont également signalé de multiples relations avec l’industrie.
Kattamis a signalé des relations financières personnelles et/ou institutionnelles avec Novartis, Bristol Myers Squib, Agios, CRISPR/Vertex, Ionis Pharmaceuticals, Vifor et Chiesi.




:quality(85):upscale()/2023/04/11/971/n/1922564/b9860d5b6435dcba0383c3.61746975_.jpg)